Epilepsy is a neurological condition that makes people
susceptible to seizures. A seizure is a change in sensation, awareness, or
behavior brought about by a brief electrical disturbance in the brain.
Seizures vary from a momentary disruption of the senses, to
short periods of unconsciousness or staring spells, to convulsions. Some people
have just one type of seizure. Others have more than one type. [1]
Epilepsy may be described basically as an intermittent
derangement of the nervous system presumably due to a sudden, excessive,
disorderly discharge of cerebral neurons. This was the postulate of Hughlings
Jackson, the eminent British neurologists of the nineteenth century, and the
modern electrophysiology offers no evidence to the contrary. The discharge
results in an almost instantaneous disturbance of sensation, loss of
consciousness, impairment of psychic function, convulsive movements, or some
combination thereof. [2]
A more common and less grave circumstance if for a seizure to
be put in an extensive series occurring over a long period of time, with most of
the attacks being more or less similar in type. In this instance they may be the
result of a burned-out lesion that originated in the past and remains as a scar.
The original disease may have passed unnoticed; or perhaps it occurred in utero,
at birth, or in infancy in parts of the brain too immature to manifest signs.
Again it may have affected a silent area in a mature brain. Patients with such
old lesions probably make up the majority of those with recurrent seizures, but
are necessarily classified as having "idiopathic epilepsy" because it is
impossible to ascertain the nature of the original disease; and the seizures may
be the only sign of brain abnormality. In this sense, all epilepsy is
"symptomatic" or "secondary" although the latter terms generally indicate that
the seizures have an identifiable and usually acquired structural cause. [2]
Seizures have been classified in many ways: according to
their supposed etiology and site of origin, on the basis of their clinical form
(generalized or focal), frequency (isolated, cyclic, prolonged, or repetitive),
or physiologic (EEG) correlates, and even in terms of their response to therapy"
The following classification is from Gastaut (1970), with the
addition of certain revisions, proposed by the Commission on Classification and
Terminology of the International League Against Epilepsy (1981). This
classification, based mainly on the clinical form of the seizure and its
electroencephalographic features, has been adopted worldwide and is generally
referred to as the International Classification"
I. Generalized seizures (bilaterally symmetrical and without
local onset)
A. Tonic or clonic or tonic - clonic (grand mal) seizures
B. Absence (petit mal)
1. Simple-loss of consciousness'" only
2. Complex-with brief tonic, clonic, or automatic movements
C. Lennox-Gastaut syndrome
D. Juvenile myoclonic epilepsy
E. Infantile spasms (West syndrome)
F. Atonic (astatic, akinetic) seizures (sometimes with
myoclonic jerks)
II. Partial (focal) seizures beginning locally
A. Simple or elementary (usually without loss of
consciousness)
1. Motor (including Jacksonian)
2. Sensory or somatosensory (visual, auditory,
olfactory, gustatory, vertiginous)
3. Autonomic
4. Psychic
B. Complex (usually with impaired consciousness:
may begin as simple partial seizure). Includes cognitive. affective,
psychosensory and psychomotor symptomatology i.e. "temporal lobe" or
"psychomotor seizures"
C. Partial seizures (simple or complex) with secondary
generalization.
III. Unilateral or predominantly unilateral seizures (tonic,
clonic, or tonic-clonic. with or without impaired consciousness)
IV. Unclassifiable (because of inadequate date) [2]
i. True petit mal
ii. Tonic – clonic major seizure (grand mal)
iii. Combined petit mal/ grand mal
iv. Primary myoclonic epilepsy
Secondary Generalized epilepsies
v. Associated with diffused brain disease,
including epilepsy secondary to specific encephalopathies and
non-specific encephalopathies (infantile spasm and Lennox – Gastaut
syndome)
Primary partial epilepsies
vi. With motor, sensorimotor, affective or
symptom or symptoms
Secondary partial epilepsies (‘lesional’)
vii. With elementary symptomatology
viii. With complex symtomatology [3]
 [14]
[14]
3. Age and epilepsy
In children, the factors of age, growth and development
exert constant influences in determining whether or not epilepsy develops.
Seizures are rare in the premature baby because of the undeveloped state of his
nervous system; however, they are more common in the full-term baby, relatively
rare in the early months of life, very common between 6 months and 4 years, and
decline in frequency until puberty. The varying convulsive threshold or
inherent susceptibility to convulse at different ages is only partly determined
by the state of anatomical maturation of the brain at the time and by the degree
of cerebral damage, if any, which is present.
There is now evidence that epilepsy involves variations of
the normal physicochemical mechanisms of cells. Epileptogenesis concerns
masses of cells in which both the threshold of stimulation is lower than normal
and the increased susceptibility to discharge can occur spontaneously when the
necessary conditioning and precipitating mechanisms develop. The resting
membrane potential of the neuronal cell has been shown to depend on the
selective permeability of the cell membrane to sodium and potassium ions. It is
highly permeable to potassium and poorly permeable to sodium. This results in a
high intracellular potassium level and a low intracellular sodium level under
normal circumstances. However, the delicate balance of the resting potentials of
cell membranes may be altered in a number of ways; for example, by changes in
the concentration of extracellular ions, by sudden mechanical or chemical
stimulation and by pathophysiological alteration of the membrane as a result of
genetic fault, disease processes or damage from whatever cause. When this
balance is upset the semipermeable characteristics alter permitting sodium and
potassium to diffuse across the membrane resulting in the ionic gradients and
their associated potentials changing. An action potential is created briefly on
the surface of the cell, and this is an effective stimulus to neighbouring
portions of the cell membrane from which the electrical potential spreads along
the axons, always away from the point of impulse generation. Synaptic
transmission involves impulse transmission in one direction from the end
branches of the axons of one neuron to the dendrites or body of another neuron.
Neurotransmitter agents are required for the passage of the impulse in this way,
and the multiple functions of the central nervous system require a highly
integrated system of excitatory and inhibitory neurotransmitter substances.
These include acetylcholine, noradrenaline, serotonin, dopamine and y-aminobutyric
acid (GABA). Enzyme mechanisms, acting on these neurotransmitters, also have an
important role.
The concept that increased permeability to ions is involved
in epileptogenicity is now accepted and it appears that all convulsions, no
matter how they are induced, result in a depletion of potassium and an increase
in sodium concentrations in neuronal cells. A defect in the active transport of
sodium and potassium appears to be closely associated with the mechanisms
involved in the generation of seizures. Derangements of the excitatory
neurotransmitter mechanisms in the central nervous system, such as
acetylcholine, are probably also involved in epileptogenesis. Derangements in
the synthesis of the powerful inhibitory substance GABA are also considered
important because they produce an alteration in the normal excitatory/
inhibitory balance of the brain towards the excitatory side. For example, there
is evidence that pyridoxal phosphate is essential for the synthesis of GABA, and
that metabolic or dietary pyridoxine deficiency may result in convulsions in
infancy. The anticonvulsant, sodium valproate, appears to act by its effect on
several enzymes involved in the synthesis and degradation of GABA.
It may be postulated, therefore, in the case of infants and
children, that not only does the anatomical maturation of the developing nervous
system play an important part in deciding when seizures will occur and what form
they will take but also that variations in the balance between inhibitory and
excitatory brain systems during maturation play a significant role in
determining the convulsive threshold or the individual's susceptibility to
convulsions at any particular age. Furthermore, nervous tissue can be rendered
hyperexcitable by changes in the body's homoeostasis acting diffusely. These
include fever, hypoxia, hypocalcaemia, hypoglycaemia, overhydration and
alterations in acid-base balance. Extraneous factors such as the administration
of convulsant drugs, the too rapid withdrawal of anticonvulsant drugs
(especially barbiturates), overdosage with various drugs producing drug toxicity
and various toxins act in a similar way on the nervous system. Emotional
disturbance, particularly when acute, and overfatigue may also act as
precipitants of seizures in those with a lowered convulsive threshold or with
established epilepsy. [3]

4. Convulsions in early infancy and infantile spasms.
In 1841, West originally reported infantile spasms as an
unusual and severe form of convulsions witnessed in his own son. Despite the
medical advances over the past century and a half, infantile spasms remain one
of the most complex and challenging conditions in pediatric neurology.
Even the terminology surrounding infantile spasms is complex,
as the term infantile spasms has been used to refer to both a specific seizure
type and a complete epilepsy syndrome. The seizure type of infantile spasms
(also referred to as epileptic spasms or simply spasms) is characterized by
brief, but often repetitive, muscle contractions usually involving the head,
trunk, and extremities. The epilepsy syndrome of infantile spasms (also known as
West syndrome) consists of the classic triad of the epileptic spasms,
hypsarrhythmia on EEG, and mental retardation.
Spasms, the seizure type, may have variable features, but
have been categorized primarily into three subtypes (flexor, extensor, and mixed
flexor-extensor) based on postural manifestations and patterns of muscle
involvement during the seizure. Flexor spasms involve flexion of the neck,
trunk, and extremities, resulting in jack-knifing at the waist and a
self-hugging motion of the arms. Extensor spasms consist of extension of the
neck, trunk, and extremities. Mixed flexor-extensor spasms involve combinations
of the above. While often confused with myoclonic or tonic seizures, spasms
represent a distinct seizure type.
Individual spasms typically last for less than 1 second to up
to 5 seconds. In many patients, spasms exhibit characteristic temporal patterns.
Fifty to eighty percent of epileptic spasms occur in clusters of 2 to more than
100 seizures. Patients may have dozens of clusters and several hundred spasms
per day, but individual variability in seizure frequency is often large.
Although spasms rarely occur during sleep, clusters of spasms are frequently
activated after awakening from sleep. Spasms are occasionally triggered by loud
noises with associated arousal from drowsiness and sleep, but are not sensitive
to photic stimulation.
Approximately one-third to one-half of patients with
epileptic spasms also have other seizure types preceding or accompanying the
onset of the spasms. Associated seizure types include partial, myoclonic, tonic,
and tonic-clonic seizures. Spasms usually cease spontaneously by age 5 but are
often replaced by other seizure types. Mental retardation and cerebral palsy
occur in about 75% and 50% respectively of children with infantile spasms. The
common combination of epileptic spasms, mental retardation, and hypsarrhythmia
on EEG constitutes the classic infantile epilepsy syndrome of infantile spasms
or West syndrome.
From an etiological standpoint, infantile spasms can be
divided into three main categories: symptomatic, cryptogenic, and idiopathic.
Because of advances in neurodiagnostic testing, symptomatic infantile spasms now
represent the largest category. The number of neurological diseases that can
result in spasms is enormous, but some of the major categories include
intrauterine insults and infections, hypoxic-ischemic encephalopathy,
malformations of cortical development, metabolic disorders, other genetic or
chromosomal defects, meningitis, tumors, and neurocutaneous syndromes.
In both cryptogenic and idiopathic infantile spasms, no
specific cause is known. However, due to the coexistence of other neurological
symptoms, especially severe developmental delay, cryptogenic cases are presumed
to have an unidentified underlying neurological disorder. Idiopathic cases of
infantile spasms are extremely rare and, by definition, consist of patients with
normal development and a good prognosis without any coexisting neurological
conditions. [6]

‘Disco Seizure’
Drawing by Tilmann Krieg [13]
5. Febrile Convulsions and status epilepticus
The well-known febrile seizure, peculiar to infants
and children between 6 months and 5 years of age (peak incidence 9 to 20 months)
and tending to be familial, is generally regarded as a benign condition. Usually
it takes the form of a single, generalized motor seizure, occurring as the
temperature rises or reaches its peak. Seldom does the seizure last longer than
a few minutes, and by the time an EEG can be obtained there is usually no
abnormality. Recovery is complete, and the risk of developing epilepsy in later
life is little or no greater than that of the general population.
This benign type of febrile seizure should not be confused
with a second and more serious type of illness in which an acute encephalitic or
encephalopathic state presents as a febrile illness with focal or prolonged
seizures, generalized or focal EEG abnormalities, and repeated episodes of
febrile convulsions with the same or different illnesses. The seizure disorder
may present as status epilepticus and the illness may end fatally; or the child
may survive and be left with mental impairment, hemiparesis, or other neurologic
abnormalities. The seizures may recur, not only with infections, but at other
times. Lennox and others have failed to separate these two types of febrile
convulsions and a third one in which an antecedent birth injury of the brain or
other disease is exposed by an episode of fever with convulsions. When cases of
all three types are lumped together under the rubric of febrile convulsions, it
is not surprising that a high percentage are complicated by atypical petit mal
and atonic and astatic spells followed by tonic seizures, mental retardation,
and psychomotor epilepsy (Lennox-Gastaut syndrome).
Other types of epilepsy are also notable with reference to
certain diseases of childhood and certain stages in the development of the
nervous system. In the young child, focal vascular or encephalitic lesion may
cause hemiparesis or other focal or lateralizing signs. Unilateral seizures
follow, and some of these are of the inhibitory type with a sudden hemiplegia
representing the seizure; or the seizure may be followed by a Todd's paralysis
lasting several hours to days. Some of these patients improve within a few
years and have no further seizures. Tumor is rarely the cause of unilateral
seizures in the child.[7]

The human electroencephalo- gram was developed
by the German psychiatrist Hans Berger
(1873-1941) in the 1920s.[13]
Primary Generalized Epilepsy
a. Grand Mal
The term convulsion is most applicable to this form of
seizure" The patient may sense its approach to anyone of several subjective
phenomena (the prodrome)" For some hours the patient may feel apathetic,
depressed, irritable, or, very rarely, the opposite-ecstatic. One or more
myoclonic jerks of the trunk or limbs, on awakening, may herald the occurrence
of a seizure later in the day" Abdominal pains or cramps, pallor or redness of
the face, throbbing headache, constipation or diarrhea have also been given
prodromal status, but we have not found them frequent enough to be helpful. In
more than half the cases there is some type of movement (usually turning of the
head and eyes or whole body), palpitation, a sinking, rising, or gripping
feeling in the epigastrium, or an unnatural sensation in another part of the
body, a few seconds before consciousness is lost. Such a remembered experience
is called the aura, which the patient regards as a sign of an impending
seizure, but which is, in reality, a simple partial seizure. The aura may
constitute the entire seizure, or it may progress, leading to unconsciousness
and a generalized seizure of the complex partial or tonic-clonic type. The aura
is important, for it may provide a clue as to the location of the "discharging
focus" or lesion"
Less often, the seizure strikes "out of the blue," i.e..
without warning, beginning with a sudden loss of consciousness and fall to the
ground. The initial motor signs are an opening of the mouth and eyes associated
with extension of the legs and abduction of the arms, flexion at the elbows and
pronation of the hands ("hands-up" position). These are followed by snapping
shut of the jaws" often with biting of the tongue, and there may be a piercing
cry as the whole musculature is seized in a spasm and air is forcibly emitted
through the closed vocal cords" Since the respiratory muscles are caught in the
tonic spasm, breathing is impossible, and after some seconds the skin and mucous
membranes become cyanotic. The bladder may empty at this stage or later, during
the postictal stupor. The pupils are dilated and unreactive to light. This is
the so called tonic phase of the seizure, and lasts for 10 to 20 sec.
There then occurs a transition from the tonic to the
clonic phase of the convulsion. At first there is a mild generalized tremor,
which is in effect a repetitive relaxation of the tonic contraction" The tremor
becomes coarser and rapidly gives way to violent muscular contractions that come
in rhythmic salvos and agitate the entire body" The eyes roll, the face becomes
violaceous and contorted by a series of grimaces" The pulse is rapid" There is a
bloody froth on the lips from the bitten tongue and excessive salivation;
rarely, periorbital hemorrhages appear. Sweating is now abundant. The clonic
jerks decrease in amplitude and frequency over a period of about 30 s, the
entire clonic phase lasting for a minute or less, as a rule. The patient remains
apneic until the end of the clonic phase, which is marked by a deep inspiration.
In the terminal phase of the seizure all movements have ended
and the patient lies still and limp in a deep coma. The pupils, equal or
unequal, now begin to contract to light. Breathing is quiet, and the skeletal
muscles are relaxed. This state persists for about 5 min, after which the
patient opens the eyes, begins to look about, and is obviously disoriented and
confused. The patient may speak and later not remember anything that was said.
If undisturbed, the patient often falls into an exhausted sleep for several
hours and may awaken with a pulsatile headache. When fully recovered, such a
patient has no memory of any part of the spell except the aura, but knows that
something has happened because of the strange surroundings (in ambulance or
hospital), the obvious concern of others, and a sore, bitten tongue and aching
muscles from the violent contractions. The contractions may even have crushed or
fractured a vertebral body, or a serious injury (fracture, subdural hematoma, or
burn) may have been sustained in the fall.
Convulsions of this type ordinarily come singly or in groups
of two or three and may occur when the patient is awake and active or during
sleep. About 5 to 8 percent of such patients will at some time have a series of
grand mal seizures without completely regaining consciousness between them. This
is called status epileptic"s and demands
urgent treatment. Conversely, instead of the whole dramatic
sequence described above, only a part of the seizure may occur. For example,
there may be only the aura without loss of consciousness, or only a brief tonic
spasm followed by a few moments of confusion, the so-called tonic seizure.
The latter type of seizure is not followed by a clonic phase and is shorter
than the tonic-clonic one. Seizures may be abbreviated by anticonvulsive
medications, and the partial motor activity may then point to the site of the
discharging lesion. [2]
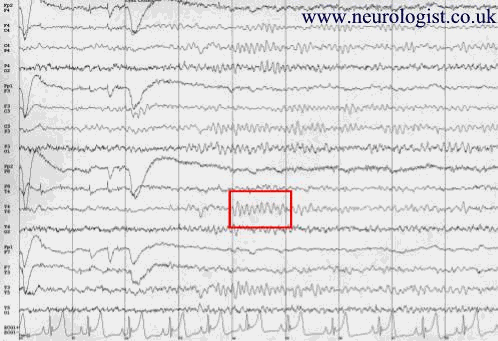
Figure 1:
This is a normal EEG showing an Alpha rhythm at 11Hz (shown in red box)
Figure 2:
 Generalised
epilepsy - "Petit Mal". The EEG shows 'spike and wave' activity occurring at 3Hz.
Generalised
epilepsy - "Petit Mal". The EEG shows 'spike and wave' activity occurring at 3Hz.
[15]
b. Petit Mal (absence seizures)
In contrast to major generalized seizures, absence or petit
mal seizures (formerly referred to as pyknoepilepsy) are notable for
their brevity and the paucity of motor activity. Indeed, they are so brief that
sometimes the patients themselves are not aware of them, and to an onlooker they
resemble a moment of absentmindedness.
The attack, coming without warning, consists of a sudden
interruption of consciousness. The patient stares and stops talking briefly or
ceases to respond. Only a few such patients (about 10 percent) are completely
motionless during the attack. Often one observes clonic movements of the
eyelids, facial muscles, or fingers or synchronous movements of both arms
occurring at a rate 013 per second, a rate that also characterizes the EEG
abnormality. Automatisms, in the form of lip smacking, chewing, and fumbling
movements of the fingers, are common during an absence attack. Lip smacking is
especially prominent in seizures induced by hyperventilation, Postural tone may
be slightly decreased or increased, and occasionally there is a mild vasomotor
disorder. As a rule such patients do not fall, and they may even continue such
complex acts as walking or riding a bicycle. After 2 to 10 s, occasionally
longer, the patient re-establishes full contact with the environment and resumes
all preseizure activity. Only a loss of the thread of conversation or the place
in reading betrays the occurrence of a momentary "blank" period (an absence).
Voluntary hypeventilation for 2 to 3 min is an effective way of inducing these
petit mal attacks.
Typical petit mal or absence is the most characteristic
epilepsy of childhood; rarely do these seizures begin before 4 years of age or
following puberty. Another attribute is their great frequency. As many as
several hundred may occur in a single day, sometimes in bursts at certain times
of the day. Most often they relate to periods of inattention and occur in the
classroom when the child is sitting quietly rather than participating actively
in his lessons. If frequent, they may disturb attention and thinking so that the
child does poorly in school. Such attacks may last for hours with no interval of
normal mental activity between them-so called petit mal or absence
status. Most cases of the latter
type have been described in adults. An attack of petit mal
status may be provoked by a borst of grand mal seizures or by the abrupt
withdrawal of anticonvulsant drugs-in which case the persistent EEG abnormality
need not be of the petit mal type. Hence the term "non convulsive generalized
status" is preferred. Petit mal may be the only type of seizure during
childhood. The attacks tend to diminish in frequency in adolescence, but rarely
do they disappear. Usually grand mal attacks become superimposed upon petit mal.
Petit Mal Variants To be distinguished from typical
absence are varieties in which the loss of consciousness is less complete or in
which myoclonus is prominent, and others in which the EEG abnormalities are less
regularly of a 3-per-second spike-and-wave type (they may be 2 to 2.5 per
second, or take the form of 4- to 6-Hz multi spike and wave complexes, or there
may be no EEG abnormalities About 30 percent of children with absence attacks
will, in addition, display symmetric or asymmetric myoclonic jerks, without loss
of consciousness. About 50 percent of such patients will also at some time have
major generalized (tonic-clonic) convulsions. A special variety of benign
myoclonic seizures occurs in late childhood and adolescence (juvenile
myoclonic epilepsy). This form of epilepsy is characterized by myoclonic
jerks affecting mainly the flexor muscles of the neck and shoulders, a tendency
to clonic-tonic-clonic seizures shortly after awakening, relatively infrequent
absence attacks, and a 4- to 6-Hz multispike and wave EEG pattern. Intelligence
is not impaired, and the response to treatment, particularly to valproate, is
excellent. The same is generally true of the other forms of epilepsy (including
"rolandic epilepsy") that have their onset in late childhood and adolescence.
In sharp contrast to the aforementioned epilepsies is a form
that has its onset in infancy and early childhond (between 2 and 6 years) and is
characterized by the occurrence of atonic (astatic) seizures, often succeeded by
various combinations of minor motor tonic-clonic and partial seizures and
intellectual impainnent, in association with a distinctive, slow (1- to
2V,-Hz) spike-and-wave EEG pattern. This is the so-called Lennox-Custaut
syndrome. Often it is preceded by infantile spasms, a characteristic EEG
picture ("hypsarrhythmia"), and an arrest in development, a triad sometimes
referred to as the West syndrome. The notion that absences, myoclonic
seizures, and akinetic seizures constitute a petit mal triad, as originally
proposed by Lennox, should be abandoned. Akinesia (motionlessness) is not unique
to any seizure type. The typical absence, with or without myoclonic jerks,
rarely causes the patient to fall and should be considered a separate entity,
because of its benignity. The early onset of atonic seizures with abrupt falls
and injuries and associated abnormalities always has grave implications, namely,
the presence of serious neurologic disease. Also, in contrast to the classical
absence, the Lennox-Gastaut syndrome may persist into adult life and is the most
difficult to treat of all forms of epilepsy. [2]
7. Myoclonic-astatic epilepsy of childhood
Lennox (1960) originally pointed out that the type of
epilepsy which occurs in a child represents a confluence of age, heredity and
structural brain abnormality. The syndrome of infantile spasms illustrates this
concept, even though heredity does not seem to play a significant role. The
heterogeneous group of disorders, now usually called the myoclonic epilepsies of
childhood, represents another set of age and development-dependent conditions
and is one in which various brain insults and heredity may be involved. The
history of the myoclonic epilepsies is a confused one. Lennox (1945) recognized
that they were distinct from true petit mal and he described their earlier time
of onset as being in the age group of 1-6 years. He noted the male predominance,
the common association with structural brain abnormality, the high incidence of
mental handicap, the variety of seizures which could occur, the fact that
hyperventilation did not provoke attacks and the lack of response to the then
specific therapy for petit mal with the dione drugs. He also described their
frequent association with an EEG pattern of slow spike-wave discharges occurring
at a rate of 1.5-2.5 complexes per second. The term 'petit mal variant' was
coined but this has since been abandoned because it tended to compound the
confusion about terminology. The term 'minor motor seizures' is also used in the
USA and in Europe. The resemblances between the clinical characteristics as
defined by Lennox (1945) and the clinical features of the infantile spasms
syndrome will be apparent. Indeed, infantile spasms are sometimes referred to as
infantile myoclonic epilepsy. Furthermore, between one- fifth and one-third of
the patients with infantile spasms go on to develop clinical myoclonic epilepsy,
with or without a seizure-free interval, and they may show the typical slow
spike-wave patterns in their EEGs.
Prior to the onset of myoclonic-astatic seizures, 84% of
affected children show normal development; the remainder show moderate
psychomotor retardation mainly affecting speech. The seizures usually begin
between 2 and 5 years of age. Boys (74%) are more often affected than girls.
The first seizure is most often a generalized tonic-clonic
seizure and rarely a myoclonic, astatic, myoclonic-astatic, or absence seizure.
Generalized tonic-clonic or clonic seizures, occur as the initial symptom in
more than half of the cases. They are usually prolonged, recurring frequently
and during the daytime. After a period of repeat generalized tonic-clonic
seizures lasting several months, so-called "minor motor seizures" appear,
consisting of myoclonic seizures, absences, and drop attacks that occur several
times a day. This period of frequent seizures lasts 1 to 3 years.
Myoclonic seizures usually involve the arms and shoulders
symmetrically and are accompanied by head nodding. The myoclonic jerks are brief
and vary in intensity: some may be so violent that the arms are flung upward;
some so mild that they are palpable rather than visible. Irregular twitching of
facial muscles, especially of the perioral and periocular musculatures, may also
be seen. A brief yell, probably a result of contraction of the diaphragm,
sometimes accompanies the myoclonic jerks.
Drop attacks may result from pure astatic, myoclonic-astatic,
or atypical absence seizures. Oguni and colleagues studied the nature of the
drop attacks with video and slow-motion analysis and found myoclonic flexor
jerks in 9 of 36 attacks, myoclonic-atonic in 2, and atonic, with or without
brief preceding events, in 25. They concluded that atonic drop attacks were a
common cause of ictal epileptic falling in myoclonic-astatic epilepsy.
Pure astatic seizures with abrupt loss of muscle tone may
occur. Astatic seizures cause either drop attacks or merely brief head nodding
or slight knee bending, depending on the extent of hypotonia. Consciousness
usually remains clear during pure astatic seizures, and the child can resume the
original posture immediately. Pure astatic seizures occur only rarely as the
only manifestation of the disorder.
The most common and characteristic seizure type, however, is
the myoclonic-astatic seizure with symmetrical myoclonic jerks immediately
followed by loss of muscle tone (postmyoclonic atonia). Lapse of consciousness
accompanied by myoclonic and/or astatic seizures occurs in 62% of the cases. The
pure myoclonic, pure astatic, or combined myoclonic-astatic seizures occur in
100% of the affected children.
Status epilepticus affects 36% of the patients with
myoclonic-astatic seizures, but the consequences are variable. It may last for
several hours or a few days without major consequences; it may last several
weeks; or it may be repeated several times during a period of 1 to 2 years.
During each episode, features of atypical absences, myoclonus, and astasia are
present in varying degrees. The child appears apathetic, hypokinetic, and
stuporous. Barely discernible myoclonic contractions and irregular twitching of
facial muscles and the hands can be detected.
Following these episodes of status, the general condition
improves, and the patient may become seizure-free after a period with only
generalized tonic-clonic seizures. However, for patients who exhibited
long-lasting episodes of status, the general condition worsens, and tonic
seizures occur during sleep and may remain as the only type of seizure the
patient experiences after the age of 10. Thus, tonic seizures are not a regular
component of the disorder. However, if they occur (usually appearing later in
the course of the disorder and during sleep), prognosis is poor.

Rolandic Epilepsy (Benign Epilepsy with Rolandic Spikes)
This type of focal motor epilepsy is unique among the partial epilepsies of
childhood in that it is a self-limited disorder and is apparently transmitted as
an autosomal dominant trait. The convulsive disorder begins between 5 and 9
years of age. It usually announces itself by a nocturnal tonic-clonic seizure
with focal onset. Thereafter the seizures take the form of clonic contractions
of one side of the face, less often of one arm or leg, and the interietal EEG
shows high voltage spikes in the lower rolandic or centrotemporal area. The
seizures are readily controlled by a single drug and gradually disappear during
adolescence.[2]
The management of epilepsy cannot be started until a correct
diagnosis has been made. Modern drug therapy is semi specific in its effects and
therefore an accurate diagnosis of the type of epilepsy from which the patient
is suffering is mandatory. Similarly, since the circumstances of each patient's
epilepsy tend to be highly individual and are important for the establishment of
a comprehensive plan of management, diagnostic study implies not only an
investigation of the type of seizure, its frequency and severity, but also a
determination of any conditioning or precipitating factors which may be present.
The identification of any localizing factors is of importance and may be
suggested by the case history, by the neurological examination or by special
investigations. (8)
A number of tests will be ordered to confirm the diagnosis of
epilepsy. These include:
Electroencephalography
Brain imaging
(CT, MRI, PET)
Blood tests
Developmental,
neurological, and behavioral tests
Electroencephalography
The most frequently used diagnostic tool for epilepsy is the
electroencephalograph (EEG). This test uses electrodes attached to the
scalp to read the brain's electrical messaging system. People who have epilepsy
often have unusual brain wave patterns even when they are not having a seizure.
EEG is most accurate, however, when it is performed within 24 hours of a
seizure.
The EEG test usually lasts about an hour and can be done in
the doctor's office or as an outpatient at the hospital. In some cases, however,
a doctor may want a 24-hour recording. Brain activity can be quite different
during sleep, and it can be helpful to measure brain waves during sleep time.
Portable EEG units can be used to monitor brain waves
throughout the day and during many different types of activities. The doctor may
also want to do video monitoring along with the EEG. Some medical centers have
special video monitoring units that help them determine whether seizures are
caused by epilepsy or by some other condition such as narcolepsy or heart
disease.
About half the people who have had an epileptic seizure have
normal EEG readings. Therefore, other diagnostic tests may be needed.
Brain Imaging
Brain imaging is often used when an adult has had a first
seizure or when a child is having convulsive seizures that are not caused by
fever.
Computerized
tomography (CT or CAT scan) and Magnetic Resonance Imaging (MRI) can be used to
reveal the structure of the brain. Both use computers to produce precise images
of the brain. These tests are useful in identifying tumors, cysts, and other
structural abnormalities.
Positron
Emission Tomography (PET) and functional MRI (fMRI) can be used to monitor the
brain's activity and to detect abnormalities in its working. These tests can
find damaged areas in the brain that are the focal points for seizures. They can
help determine whether a patient is a good candidate for surgery and can be used
to guide surgery.
Blood Tests
Doctors use blood tests to screen for metabolic or genetic
disorders that may be linked to seizures and to look for other conditions that
may cause seizures, such as infections, lead poisoning, anemia, or diabetes.
Metabolic and genetic screening is most often done when examining a child.
Developmental, Neurological, And
Behavioral Tests
Tests to measure coordination and muscle control, behavior,
and intellectual capacity can help find out what is causing epilepsy and how the
seizures may be affecting the patient. (12)

10. The multidisciplinary approach
Because of the complexities of diagnosis and management of
epilepsy, the question`of the need for special centres and clinics for epilepsy
needs consideration and has been reviewed in detail by Brown (1976). As he has
pointed out, there has been a veritable explosion of knowledge about the
genetics, electrophysiology, neurochemistry and pharmacology of epilepsy in
recent years making it difficult for anyone but the specialist to keep abreast
of the changing epileptic scene. There has been a remarkable increase in
knowledge of the adverse effects of anticonvulsant drugs, including the
recognition of various clinical syndromes, making it imperative that the
physician dealing with epilepsy should have access to facilities for monitoring
the levels of these drugs in the blood plasma. Something approaching a third of
all epileptic children are mentally retarded to a greater or lesser degree and
many have behavioral difficulties. These include the hyperkinetic behavior and
other difficulties of the child with temporal lobe epilepsy, and the withdrawn
autistic behavior of the child who has had infantile spasms or severe myoclonic
epilepsy and also a host of less severe reactions including enuresis, encopresis,
phobias and anxieties. Medico social problems are also common in the family with
an epileptic child - the predicaments of the patient and his family. It is clear
that the child with epilepsy needs a multidisciplinary
approach but, because of the way in which the growth and development of
the child are so intimately linked with the type of epilepsy which may afflict
him, in contrast to the relatively unchanging state of the adult, it seems
unlikely that special centers for epilepsy are the answer as far as paediatric
epileptology is concerned. The multidisciplinary approach to epilepsy in
childhood should involve the participation of people with many different skills
and interests, but not necessarily working under the same roof. The family
doctor will have the closest contact with the family in their predicament, and
the pediatrician will be involved when the child is first referred with
epilepsy. The pediatrician is in the front line as far as the prevention of
epilepsy. The community physician, the teacher, the child psychiatrist and the
psychologist may all be involved. A special outpatient clinic for children with
epilepsy, based in a children's hospital or in the children's department of a
general hospital and under the supervision of a pediatrician with a special
interest in epilepsy or of a pediatric neurologist, is particularly valuable
because it allows time to be allotted to the parents for discussion of their
problems and also because it ensures continuing care by one concerned physician.
The place of special schools for children with epilepsy has already been
mentioned. Finally, there is a need for a few highly specialized 'centers of
excellence' where intractable and difficult cases can be studied in detail using
all available modern techniques, and where the psychotherapeutic management of
the whole family in their predicament can be undertaken.(9)

11. Treatment of Epilepsy
Epilepsy is usually treated with anti-epileptic medications.
Approximately 70% of people with epilepsy have either complete or very good
seizure control with medication. About 30% of people with epilepsy cannot
control their seizures with medicine. Treatment options for these patients
include epilepsy surgery, vagus nerve stimulation, and the ketogenic diet (used
in children).
Once the exact seizure type and
epilepsy syndrome has been determined, the doctor usually chooses the best
medication effective for that type of seizure. Several other factors are
considered such as cost, dosing schedule, patient age, previous drug
interactions, other existing health conditions, and child bearing potential.
Monotherapy is the ideal type of therapy to manage side effects and dosing more
easily. Approximately 50% of patients are free of seizures with one medication.
It may be necessary to add a second drug if one drug does not control your
seizures. The goal is to control the seizures with the fewest possible side
effects.
Common anti-epileptic medications:
Phenytoin (Dilantin)
is one of the oldest and most commonly used drug to treat seizures. It is
most effective against tonic-clonic seizures or complex partial seizures.
Phenobarbital is a barbiturate used to treat tonic-clonic and simple partial
seizures. It was the first drug to be used to treat epilepsy, and it is
known to cause drowsiness. It is rarely used to treat new cases of epilepsy.
Valproic
acid (Depakote, Depakene) is used for partial and tonic/clonic seizures.
Carbamazepine (Tegretol, Carba-trol) is another drug to treat some types of
seizures. It’s also used to treat pain caused by trigeminal neuralgia.
Since 1992, eight new medications have been approved
primarily as a supplemental therapy to treat epilepsy:
Felbamate (Felbatol)
is restricted to patients with severe intractable partial and secondary
generalized seizures and in patients with Lennox-Gastaut syndrome who have
not responded to other medication.
Gabapentin (Neurontin)
is used for partial seizures with or without secondary generalization.
Lamotrigine
(Lamictal) is used for partial seizures and in generalized seizures of
Lennox-Gastaut syndrome in children and adults.
Topiramate (Topamax)
is used in adults with partial onset and generalized tonic-clonic seizures.
Tiagabine (Gabitril)
is used in adults and children more than 12 years old with partial seizures.
Levetiracetam (Keppra) is used for partial onset seizures.
Oxcarbazepine (Trileptal) is used in adults and children 4-16 years old with
partial seizures.
Zonisamide (Zonegran)
is used for partial seizures.
The new formulations and drug delivery systems include an
extended-release form of carbamazepine, fosphenytoin, an intravenous formulation
of valproic acid, and diazepam rectal gel.
As with all drugs, there are side effects and drug
interactions. Most common side effects include fatigue, drowsiness, nausea, gum
problems, weight gain and blurred vision. If the patient has unacceptable side
effects, changing to another medication may improve these adverse effects. Also,
these drugs may reduce the effectiveness of birth control pills. If the patient
is pregnant or thinking about getting pregnant, she should not take any
anti-seizure medications that are known to cause birth problems. The patient
needs to discuss this with her physician.

Surgical treatments
When medication fails or the side effects become
unacceptable, surgical treatments may be used.
Vagus nerve stimulation offers a unique
treatment option and may help the patient if his seizures cannot be
controlled with medications or if the patient experiences unacceptable side
effects from medication. It may also be used if he is not a candidate for
surgery. The pacemaker-like device is implanted in the patient’s chest with
a wire that is wrapped around the vagus nerve, which is located in the neck.
The stimulator applies a small adjustable electrical current to the vagus
nerve. Some minor side effects can be experienced, such as a tingling or
change in voice during stimulation. In addition, he might need to place a
magnet over the device and provide additional stimulation to possibly stop a
seizure in progress. The success rate of vagus nerve stimulation is
comparable to the newer medications. VNS is not a cure for seizures. It is
important to decide if epilepsy surgery is a better option for the patient
before the stimulator is implanted.
Epilepsy surgery
may be a treatment option if the
seizures that cannot be
controlled
by medications. Most commonly, a portion of the brain that generates
seizures is removed during the surgical procedure. In addition to operations
that remove a small part of the brain where seizures begin, other procedures
may be done to interrupt the spread of electrical energy/discharges in your
brain. Surgery can be considered, only if the seizures that arise from one
focal area of the brain or that can be isolated from the other parts of the
brain. Depending on the site of seizure onset and the underlying cause,
50-80% of patients who have epilepsy surgery can become seizure free.
Surgery is especially beneficial for those with temporal lobe epilepsy.


Ketogenic diet
A ketogenic (high-protein/fat) diet may be a treatment option
for children who have many side effects from anti-seizure drugs, or whose
seizures cannot be controlled by them. This special restricted-calorie diet
tricks the body into burning fat, instead of glucose, for energy. The diet is
very high in fats and low in protein and carbohydrates. It produces a change in
the body's chemistry called ketosis, which has the effect of controlling
seizures or reducing their frequency, in two of three children placed on the
diet. Although not all children benefit, parents report that children who do
benefit are more alert and active than they were previously.
The ketogenic diet must be established by a dietitian,
weighed out in grams by the family, and followed by a doctor just as if it were
a course of drug treatment. Like other treatments, the ketogenic diet has some
side effects, which are monitored through blood and urine tests and follow-up
visits. The diet is used primarily to treat children for a limited period of
time, after which the diet may be slowly tapered and regular food slowly
reintroduced. If seizures return, the diet may be re-instituted. Studies are
underway to see whether the ketogenic diet may also work for some adults. Early
results suggest that it may, but the long-term effects of such a high-fat diet
are unknown.
Clinical trials
Clinical trials are research studies in which new treatments
- drugs, diagnostics, procedures, vaccines, and other therapies - are tested in
people to see if they are safe and effective. Research is always being conducted
to improve the standard of medical care and explore new drug and surgical
treatments. [10]

Lord Byron with Albanian costume
(Thomas Phillips, 1813)
Did Lord Byron really have
epilepsy? If one searches through the biographies of the greatest English
Romantic poet - either to find evidence to support or prove wrong Nietzsche's
opinion - one finds passages in a few places which suggest that Byron might
possibly have suffered epileptic seizures.[13]
12. Psychosocial Effects of Epilepsy
Many people report that the most
frightening thing about seizures is their
unpredictability.
Children with epilepsy must learn to live with epilepsy.
Epilepsy is a chronic disorder and may have the same types of
effects on children as would a chronic disease.
Epilepsy is episodic; in other words, no one can predict when
a seizure will occur. Therefore, it may be even more difficult to adapt to
epilepsy than to other more predictable chronic conditions.
Even a child whose epilepsy is well controlled with
medication may still be fearful about having another seizure, especially in the
presence of peers.
It is not enough to treat only the medical aspects of the
child's seizure disorder. The psychosocial, emotional and physical components of
the child's life must all be considered as well. Research shows that parents may
interfere with their child's healthy psychosocial adjustment by being
overprotective, rejecting, or having low expectations of him/her. Children with
epilepsy can attain a high quality of life. Parents who know when to let their
children have their own freedom, who accept and support their children as they
are, and who encourage their children to have new experiences can greatly assist
their children in attaining the skills which will help them to have fulfilling
lives.

Research shows that the attitudes of others about epilepsy
has more impact on people than does epilepsy itself. Attitudes toward people
with an invisible impairment, such as epilepsy, are generally less positive than
towards those with a visible impairment. The social stigmas associated with
epilepsy can be very detrimental to children with epilepsy. Children are often
very self-conscious about their appearance and it may be very difficult for them
to deal with having even brief seizures in public. Children fear being viewed as
"different."
A newly diagnosed child's perceptions of the disorder largely
depend on the parents' reactions to it. How the child's parents deal with the
disorder, rather than the severity or frequency of the child's seizures,
determines how well the child adapts to the disorder. Instill confidence in the
child by praising what s/he can do. Allow the child to make choices to foster
independence. Do not isolate the child, saying "It's for your own good." All
children deserve the same opportunities. All children need to learn how to take
reasonable risks. Facing fear and failure is a valuable learning experience
necessary for a child to grow and mature.
The social stigma of epilepsy is experienced sooner or later
by both children and adults. Many people feel anxious about the prospect of
having to deal with a person who has epilepsy.
To help build your child's self esteem, you should encourage
openness. Secrecy reinforces the idea that epilepsy may be something shameful.
Secrecy interferes with acceptance and can erode a child's feelings of
self-worth. A straightforward approach to dealing with epilepsy may appear to be
difficult initially but will be more helpful in the long run.
The decision whether or not to tell others about a child's
epilepsy depends on many factors. Before you tell anyone outside of the family
about the child's condition, the parents should discuss it with the child.
Ensure that the child understands why it is necessary to disclose his/her
condition. Allow your child to play a role in deciding who to tell about his/her
condition.
Children and Self-Esteem
Self-esteem may be the single most important attribute for a
child to develop. For children with epilepsy, developing self-esteem can be a
challenge, as society often holds negative attitudes toward epilepsy.
It is important for adults to focus on what is within their
control. Parents can instill feelings of self-acceptance and self-worth within
their children. In order to help the child to develop self-esteem, the parents
must accept and acknowledge their child's feelings. (This also means that
parents must come to acknowledge and accept their own feelings. Shame develops
when parents tell their child not to feel a certain way, or when they find their
child's emotions unacceptable.
For a child with a chronic disorder such as epilepsy, the
development of self-esteem may be a great challenge. At times, the child may
struggle with a low self image and increased feelings of anxiety and loss of
control due to the unpredictable nature of seizures.
Just as parents have a multitude of concerns about their
child's seizures, the child may also be coping with a myriad of feelings,
including fear, embarrassment, anger, denial and anxiety. These feelings may
come or go, and may fluctuate in duration and intensity. Uncertainty about when
a seizure will occur, fear of death, fear of medical tests, fear that kids will
tease are feelings that can follow some children into adult years.
If parents are comfortable with their child's seizure
disorder, it will help their child to be more comfortable with the disorder. If
parents are ashamed of or anxious about their child's epilepsy, then their child
will be ashamed or anxious too. Fostering self acceptance, self-confidence and
self worth helps to prepare your child to deal with his/her own feelings and the
attitudes of others.
Sharing information about epilepsy in words that the child
understands will help to remove some of the mystery surrounding the seizures.
Concealing information about epilepsy in an attempt to protect a child is not
helpful. Children often fill in the blanks with misinformation which can create
unfounded concerns.
The child's self image, in large part, is affected by the
reactions of family, friends and involved medical professionals. The parents,
muat be able to educate others, inform family and friends that the seizures are
a temporary interruption to the child's day. It is important for others to
understand that the child is a child first, who simply happens to have seizures.
Psychosocial Effects of Epilepsy on Parents
A single seizure — especially the child's first seizure — usually has its
greatest effects on the parent, not the child.
Parents often go through a variety of stages after
finding out that their child has
epilepsy.
The first emotion most people experience is fear. Parents
fear the unknown most of all. Because epilepsy is episodic, there is no way of
knowing when or if another seizure will occur. Grief is the next emotion that
parents often feel. Parents grieve for the child they think is no longer the
same as before, for the effects they think epilepsy will have, and for how
epilepsy will interfere with all of their lives. Parents need to put their grief
into perspective. Finding support groups and other resources can help parents
accept their child's condition. Eventually, grieving must come to an end and
parents must find a more productive way to deal with epilepsy.
Following grief, parents are often angry. They often wonder, "Why did my child
get epilepsy?" They may feel anger towards the medical staff for not
doing/knowing more. Anger is not a productive emotion; instead parents should
try to discuss their feelings or ideas with others. It is important to realize
that having these feelings is normal and that they will usually pass with time.
Many people believe that the single biggest issue for parents
raising children with epilepsy is overprotection. The effects of overprotection
on children can be serious and long lasting. They may include dependency,
hypochondria, low self-esteem, underachievement and immaturity. Parents should
not let their anxiety about epilepsy control their life or that of their child.
Parents should be a bit more cautious but should not let the fear of another
seizure run your life.
Psychosocial Effects of Epilepsy on Children
Children may also go through the same emotional stages as
their parents. Children may fear dying, fear losing control, fear the
unpredictability of the disorder.
Children also grieve, as they may be forced to change what they once could do.
Therefore, it is important that when restrictions are put in place, parents find
other activities that the child can engage in safely. It may be helpful for
children with epilepsy to meet other children with epilepsy.
For all children, self-esteem is crucial in their accepting
themselves as they are.
Parents who tell their child that the child's medication is a
vitamin, or just something that is good for them, have not accepted their
child's epilepsy and are not allowing their child to begin to accept the
disorder. Parents are encouraged to let their children be responsible for taking
their own medication (still with supervision of the younger child) so that the
child feels that they can take control of their epilepsy.
Teachers can be very helpful in informing parents of any
changes in the performance or personality of their child (such as side effects
related to drug toxicity). Children are extremely motivated towards successful
adjustment.
The behavioural difficulties that children with epilepsy tend
to experience are generally a result of the frightening, helpless and traumatic
state in which children with epilepsy often find themselves. Seizures are often
experienced as attacking and threatening, which in turn heightens the child's
feelings of vulnerability. Having to deal with the unpredictable, attacking
nature of seizures is often an extremely frightening and tense experience for
children. Children with epilepsy may feel vulnerable about their bodies and
fragile about their well being.
While children with epilepsy may exhibit differences in their
physical and neurological development, they must still achieve the same
emotional and physical milestones as all other children.
Having epilepsy can create an enormous challenge for the
child developing adaptive behaviour. Children work very hard to make a positive
adjustment to epilepsy.
However, having to deal with seizures as part of their everyday development has
the potential to traumatize children. Despite the best efforts of parents,
physicians, counsellors, etc., the child may feel so overwhelmed that their
psychosocial development is affected. When trying to cope with the disruption of
epilepsy, the child may experience behavioural and emotional (psychosocial)
difficulties. Even when seizures are controlled, some children still have these
difficulties.
Warning signs, especially when seen in combination, may indicate that your child
is having trouble dealing with epilepsy. These warning signs may include
hyperactivity, depression, confusion, disorganized speech, inability to listen
to and comply with directions, and lack of pleasure.
Children with epilepsy often tend to have more difficulties
in school. Usually, this is not a direct result of epilepsy but due to the
consequences of fear and anxiety which children with epilepsy experience. Many
children with epilepsy do not have learning problems.
The information and explanations that parents give to their
children act to buffer their child's experiences and build the foundation of
their future awareness. Complications can occur when children's feelings are not
acknowledged or comforted. Parents must not allow their feelings to make things
worse by preventing their child from engaging in various experiences or having
independence.
Establishing routines for doctors visits, taking medications,
having EEGs, etc., can help the child accept and deal with having epilepsy. In
middle childhood and early adolescence, children have new concerns regarding
their medical condition: these may require the child to gain a new level of
knowledge and understanding about epilepsy and how it will impact on their life
as they grow. Older children often need to sort through their feelings about
epilepsy before they can move on and accept their condition.
Depression is quite common in children who have epilepsy. It may be a side
effect of antiepileptic medication, or it may be caused by other factors
including stress, major disappointment, or a chemical imbalance in the body.
Depression can have a major impact on your child's life.
It is important for parents and teachers to be aware of the possibility that the
child with epilepsy may experience depression. This should not be surprising
since the diagnosis of epilepsy can be quite overwhelming for a child. The child
who is depressed is unlikely to talk to others about how they are feeling.
The following signs may indicate the child is depressed:
o
Emotional Changes. The child may exhibit a troubled and unhappy state of
mind. The child may feel unhappy, worried, guilty, angry, fearful, helpless,
lonely or rejected. The child may even contemplate suicide. Look for the usually
happy and active child who suddenly becomes quiet and withdrawn.
o
Intellectual Changes. The child may exhibit trouble concentrating or may
experience chronic negative thoughts. Look for the child who was a good student,
but suddenly receives bad marks.
o
Physical
Changes. The child may complain of headaches, or aches and pains. The child
may also appear to be unmotivated, with a lack of energy and sleepy all of the
time. The child may also develop eating problems.
o
Behavioural Changes. The child may withdraw from interacting with others.
The child may cry easily and display angry or aggressive behaviour. The child
may not want to interact with friends, play or take part in fun activities that
s/he used to enjoy.
If the any of the above signs are noticed, the child should be encouraged to
discuss his/her feelings with his/her parents, another caregiver or another
trusted individual.
If these warning signs are noticed by a teacher, the teacher
should report them to
the child's parents. A school counsellor may be able to help.
Professional medical help should also be sought. It is
important for the child's physician to determine whether there is a physical
cause for the child's headaches, aches and pains, sleepiness, etc.
Depression is treatable and it can be overcome. [11]
By writing this essay I tried to approach Epilepsy in Childhood from a few
different angles. Some of the causes and types of seizures have been described
and some were left out. A lot can be said and written about this condition. My
personal view is that the cause or the kind of syndrome a patient/child has is
not so important. What it matters most is each individual patient/ child. Each
case has to be individualized. Each patient is a human being and not a machine
that "has to be fixed". The most important aspect of this condition and, I
believe, of any condition, is the psychosocial approach and how each patient
feels and copes with the condition.
 [13]
[13]
|
a References
b |
| No |
|
|
Epilepsy
Foundation http://www.epilepsyfoundation.org/ |
|
RaymondD.Adams,
Maurice Victor. Principles of Neurology, Fourth Edition.1989; 249-271
|
|
Nial V.
O’Donohoe. Epilepsies of Childhood, Second Edition. 1985;6 |
|
RaymondD.Adams,
Maurice Victor. Principles of Neurology, Fourth Edition.1989; 249-271
|
|
Fritz Dreifuss
and Oliver Dulac, Myoclonic astatic epilepsy of Childhood.1993 |
|
Dr Michael
Wong. Pediatric Epilepsy Center, Washington University School of Medicine |
|
RaymondD.Adams,
Maurice Victor. Principles of Neurology, Fourth Edition.1989; 249-271
|
|
Nial V.
O’Donohoe. Epilepsies of Childhood, Second Edition. 1985; 9 |
|
Nial V.
O’Donohoe. Epilepsies of Childhood, Second Edition. 1985;189 |
|
Epilepsy.
Mayfield Clinic, http://mayfieldclinic.com/ |
|
Children
Living with Epilepsy. Epilepsy Ontario. http://epilepsyontario.org |
|
Your Medical
Source. http://yourmedicalsource.com |
|
Epilepsy and
Art: Votive tables, paintings, sculptures. German Epilepsy Museum http://www.epilepsiemuseum.de/ |
|
Patrick
Gaston’s webpage. http://studentweb.tulane.edu/~pgaston/ |
|
http://neurologist.co.uk |

Epileptic girl suffering a seizure.
Votive tablet from 1766

‘The Epileptic Mind’
What doubtless remained longer than leprosy, and would
persist when the lazar houses had been empty for years, were the values and
images attached to the figure of the leper as well as the meaning of his
exclusion, the social importance of that insistent and fearful figure which was
not driven off without first being inscribed within a sacred circle."
Michel Foucault, Madness and Civilization:
a history of Insanity in the age of Reason
By Panicos Shangaris
RCSI, March 2005
panicos@shangaris.com







